
RNA-seq 保姆教程:差异表达分析(二)
创始人
2024-02-04 01:57:14
介绍
RNA-seq 目前是测量细胞反应的最突出的方法之一。RNA-seq 不仅能够分析样本之间基因表达的差异,还可以发现新的亚型并分析 SNP 变异。本教程[1]将涵盖处理和分析差异基因表达数据的基本工作流程,旨在提供设置环境和运行比对工具的通用方法。由于完整版过长,因此分为两部分,需要获取完整版的,请跳转文末。
7. 差异分析
将基因计数导入 R/RStudio
工作流程完成后,您现在可以使用基因计数表作为 DESeq2 的输入,使用 R 语言进行统计分析。
7.1. 安装R包
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2") ; library(DESeq2)
biocLite("ggplot2") ; library(ggplot2)
biocLite("clusterProfiler") ; library(clusterProfiler)
biocLite("biomaRt") ; library(biomaRt)
biocLite("ReactomePA") ; library(ReactomePA)
biocLite("DOSE") ; library(DOSE)
biocLite("KEGG.db") ; library(KEGG.db)
biocLite("org.Mm.eg.db") ; library(org.Mm.eg.db)
biocLite("org.Hs.eg.db") ; library(org.Hs.eg.db)
biocLite("pheatmap") ; library(pheatmap)
biocLite("genefilter") ; library(genefilter)
biocLite("RColorBrewer") ; library(RColorBrewer)
biocLite("GO.db") ; library(GO.db)
biocLite("topGO") ; library(topGO)
biocLite("dplyr") ; library(dplyr)
biocLite("gage") ; library(gage)
biocLite("ggsci") ; library(ggsci)
7.2. 导入表达矩阵
开始导入文件夹中的 featureCounts表。本教程将使用DESeq2对样本组之间进行归一化和执行统计分析。
# 导入基因计数表
# 使行名成为基因标识符
countdata <- read.table("example/final_counts.txt", header = TRUE, skip = 1, row.names = 1)
# 从列标识符中删除 .bam 和 '..'
colnames(countdata) <- gsub(".bam", "", colnames(countdata), fixed = T)
colnames(countdata) <- gsub(".bam", "", colnames(countdata), fixed = T)
colnames(countdata) <- gsub("..", "", colnames(countdata), fixed = T)
# 删除长度字符列
countdata <- countdata[ ,c(-1:-5)]
# 查看 ID
head(countdata) # 如下图
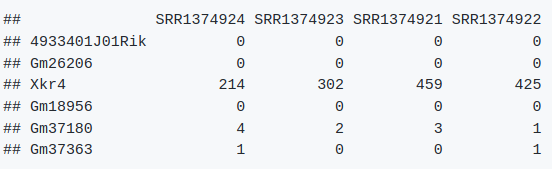
7.3. 导入metadata
导入元数据文本文件。 SampleID必须是第一列。
# 导入元数据文件
# 使行名称与 countdata 中的 sampleID 相匹配
metadata <- read.delim("example/metadata.txt", row.names = 1)
# 将 sampleID 添加到映射文件
metadata$sampleid <- row.names(metadata)
# 重新排序 sampleID 以匹配 featureCounts 列顺序。
metadata <- metadata[match(colnames(countdata), metadata$sampleid), ]
# 查看 ID
head(metadata) # 如下图
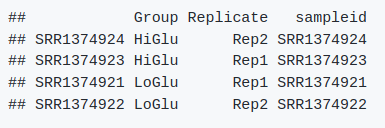
7.4. DESeq2对象
根据计数和元数据创建 DESeq2对象
# - countData : 基于表达矩阵
# - colData : 见上图
# - design : 比较
ddsMat <- DESeqDataSetFromMatrix(countData = countdata,
colData = metadata,
design = ~Group)
# 查找差异表达基因
ddsMat <- DESeq(ddsMat)
7.5. 统计
获取基因数量的基本统计数据
# 使用 FDR 调整 p-values 从检测中获取结果
results <- results(ddsMat, pAdjustMethod = "fdr", alpha = 0.05)
# 结果查看
summary(results) # 如下图
# 检查 log2 fold change
## Log2 fold change is set as (LoGlu / HiGlu)
## Postive fold changes = Increased in LoGlu
## Negative fold changes = Decreased in LoGlu
mcols(results, use.names = T) # 结果如下

8. 注释基因symbol
经过比对和总结,我们只有带注释的基因符号。要获得有关基因的更多信息,我们可以使用带注释的数据库将基因符号转换为完整的基因名称和 entrez ID 以进行进一步分析。
收集基因注释信息
# 小鼠基因组数据库
library(org.Mm.eg.db)
# 添加基因全名
results$description <- mapIds(x = org.Mm.eg.db,
keys = row.names(results),
column = "GENENAME",
keytype = "SYMBOL",
multiVals = "first")
# 添加基因 symbol
results$symbol <- row.names(results)
# 添加 ENTREZ ID
results$entrez <- mapIds(x = org.Mm.eg.db,
keys = row.names(results),
column = "ENTREZID",
keytype = "SYMBOL",
multiVals = "first")
# 添加 ENSEMBL
results$ensembl <- mapIds(x = org.Mm.eg.db,
keys = row.names(results),
column = "ENSEMBL",
keytype = "SYMBOL",
multiVals = "first")
# 取 (q < 0.05) 的基因
results_sig <- subset(results, padj < 0.05)
# 查看结果
head(results_sig) # 如下图
将所有重要结果写入 .txt 文件
# 将归一化基因计数写入 .txt 文件
write.table(x = as.data.frame(counts(ddsMat), normalized = T),
file = 'normalized_counts.txt',
sep = '\t',
quote = F,
col.names = NA)
# 将标准化基因计数写入 .txt 文件
write.table(x = counts(ddsMat[row.names(results_sig)], normalized = T),
file = 'normalized_counts_significant.txt',
sep = '\t',
quote = F,
col.names = NA)
# 将带注释的结果表写入 .txt 文件
write.table(x = as.data.frame(results),
file = "results_gene_annotated.txt",
sep = '\t',
quote = F,
col.names = NA)
# 将重要的注释结果表写入 .txt 文件
write.table(x = as.data.frame(results_sig),
file = "results_gene_annotated_significant.txt",
sep = '\t',
quote = F,
col.names = NA)
9. 绘图
有多种方法可以绘制基因表达数据。下面只列出了一些流行的方法。
9.1. PCA
# 将所有样本转换为 rlog
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
# 按列变量绘制 PCA
plotPCA(ddsMat_rlog, intgroup = "Group", ntop = 500) +
theme_bw() +
geom_point(size = 5) +
scale_y_continuous(limits = c(-5, 5)) +
ggtitle(label = "Principal Component Analysis (PCA)",
subtitle = "Top 500 most variable genes")
9.2. Heatmap
# 将所有样本转换为 rlog
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
# 收集30个显著基因,制作矩阵
mat <- assay(ddsMat_rlog[row.names(results_sig)])[1:40, ]
# 选择您要用来注释列的列变量。
annotation_col = data.frame(
Group = factor(colData(ddsMat_rlog)$Group),
Replicate = factor(colData(ddsMat_rlog)$Replicate),
row.names = colData(ddsMat_rlog)$sampleid
)
# 指定要用来注释列的颜色。
ann_colors = list(
Group = c(LoGlu = "lightblue", HiGlu = "darkorange"),
Replicate = c(Rep1 = "darkred", Rep2 = "forestgreen")
)
# 使用 pheatmap 功能制作热图。
pheatmap(mat = mat,
color = colorRampPalette(brewer.pal(9, "YlOrBr"))(255),
scale = "row",
annotation_col = annotation_col,
annotation_colors = ann_colors,
fontsize = 6.5,
cellwidth = 55,
show_colnames = F)
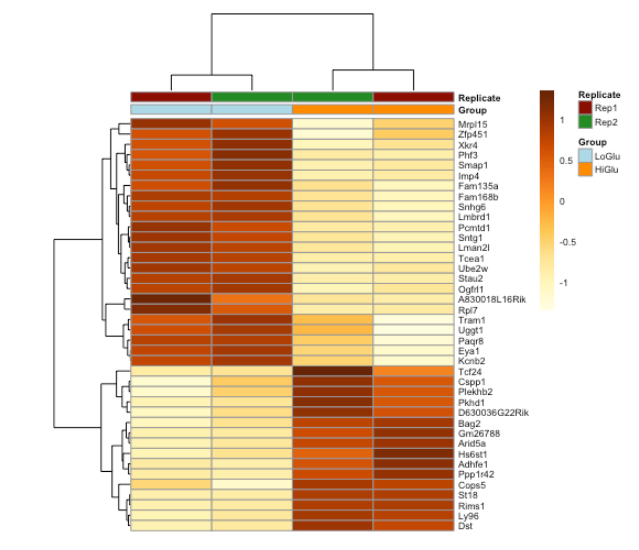
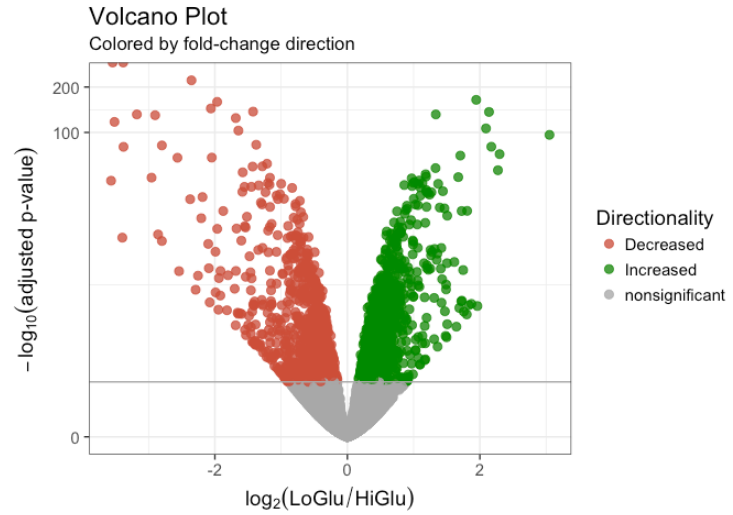
9.3. Volcano
# 从 DESeq2 结果中收集倍数变化和 FDR 校正的 pvalue
## - 将 pvalues 更改为 -log10 (1.3 = 0.05)
data <- data.frame(gene = row.names(results),
pval = -log10(results$padj),
lfc = results$log2FoldChange)
# 删除任何以 NA 的行
data <- na.omit(data)
## If fold-change > 0 and pvalue > 1.3 (Increased significant)
## If fold-change < 0 and pvalue > 1.3 (Decreased significant)
data <- mutate(data, color = case_when(data$lfc > 0 & data$pval > 1.3 ~ "Increased",
data$lfc < 0 & data$pval > 1.3 ~ "Decreased",
data$pval < 1.3 ~ "nonsignificant"))
# 用 x-y 值制作一个基本的 ggplot2 对象
vol <- ggplot(data, aes(x = lfc, y = pval, color = color))
# 添加 ggplot2 图层
vol +
ggtitle(label = "Volcano Plot", subtitle = "Colored by fold-change direction") +
geom_point(size = 2.5, alpha = 0.8, na.rm = T) +
scale_color_manual(name = "Directionality",
values = c(Increased = "#008B00", Decreased = "#CD4F39", nonsignificant = "darkgray")) +
theme_bw(base_size = 14) +
theme(legend.position = "right") +
xlab(expression(log[2]("LoGlu" / "HiGlu"))) +
ylab(expression(-log[10]("adjusted p-value"))) +
geom_hline(yintercept = 1.3, colour = "darkgrey") +
scale_y_continuous(trans = "log1p")
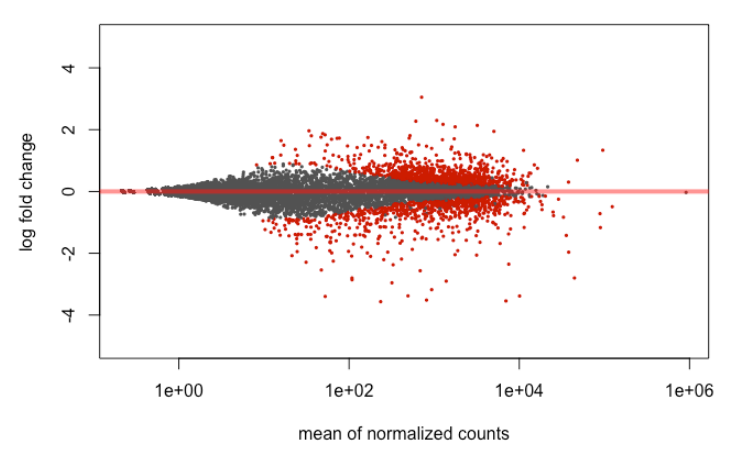
9.4. MA
plotMA(results, ylim = c(-5, 5))
9.5. Dispersions
plotDispEsts(ddsMat)
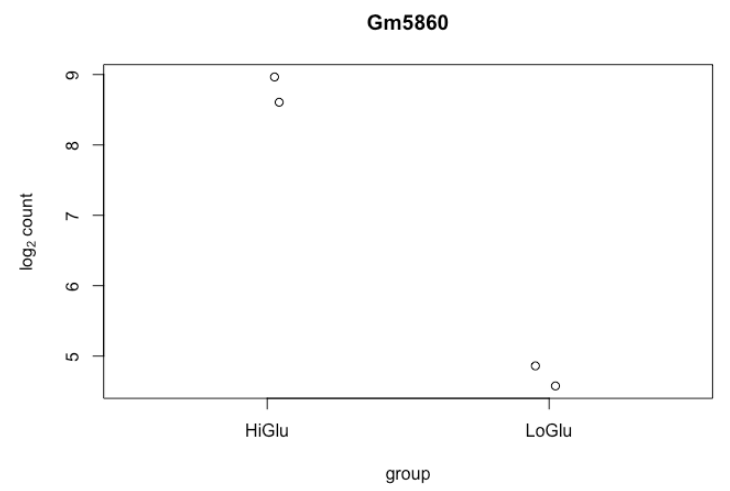
9.6. 单基因图
# 将所有样本转换为 rlog
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
# 获得最高表达的基因
top_gene <- rownames(results)[which.min(results$log2FoldChange)]
# 画单基因图
plotCounts(dds = ddsMat,
gene = top_gene,
intgroup = "Group",
normalized = T,
transform = T)
10. 通路富集
从差异表达基因中寻找通路
通路富集分析是基于单个基因变化生成结论的好方法。有时个体基因的变化是难以解释。但是通过分析基因的通路,我们可以收集基因反应的视图。
设置矩阵以考虑每个基因的 EntrezID 和倍数变化
# 删除没有任何 entrez 标识符的基因
results_sig_entrez <- subset(results_sig, is.na(entrez) == FALSE)
# 创建一个log2倍数变化的基因矩阵
gene_matrix <- results_sig_entrez$log2FoldChange
# 添加 entrezID 作为每个 logFC 条目的名称
names(gene_matrix) <- results_sig_entrez$entrez
# 查看基因矩阵的格式
##- Names = ENTREZ ID
##- Values = Log2 Fold changes
head(gene_matrix) # 如下图
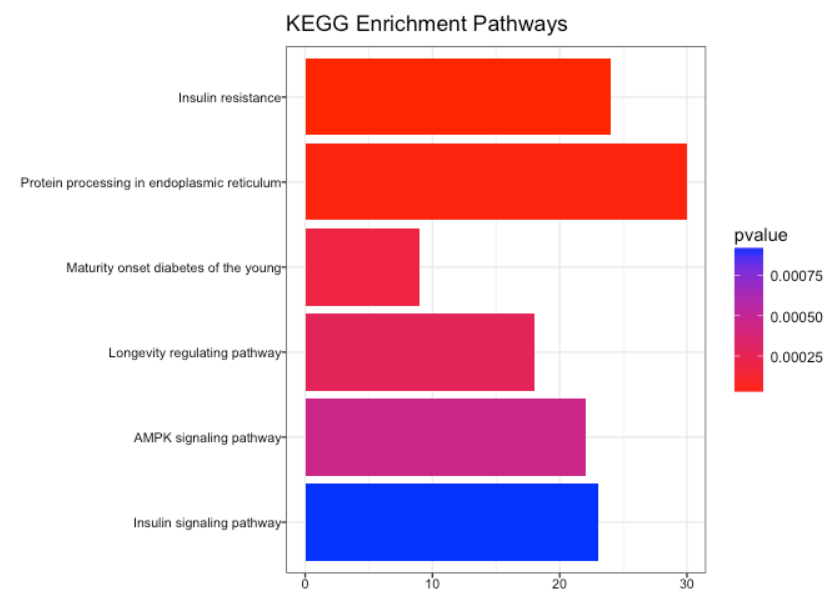
10.1. KEGG
使用 KEGG数据库丰富基因
kegg_enrich <- enrichKEGG(gene = names(gene_matrix),
organism = 'mouse',
pvalueCutoff = 0.05,
qvalueCutoff = 0.10)
# 结果可视化
barplot(kegg_enrich,
drop = TRUE,
showCategory = 10,
title = "KEGG Enrichment Pathways",
font.size = 8)
10.2. GO
使用 Gene Onotology丰富基因
go_enrich <- enrichGO(gene = names(gene_matrix),
OrgDb = 'org.Mm.eg.db',
readable = T,
ont = "BP",
pvalueCutoff = 0.05,
qvalueCutoff = 0.10)
# 结果可视化
barplot(go_enrich,
drop = TRUE,
showCategory = 10,
title = "GO Biological Pathways",
font.size = 8)
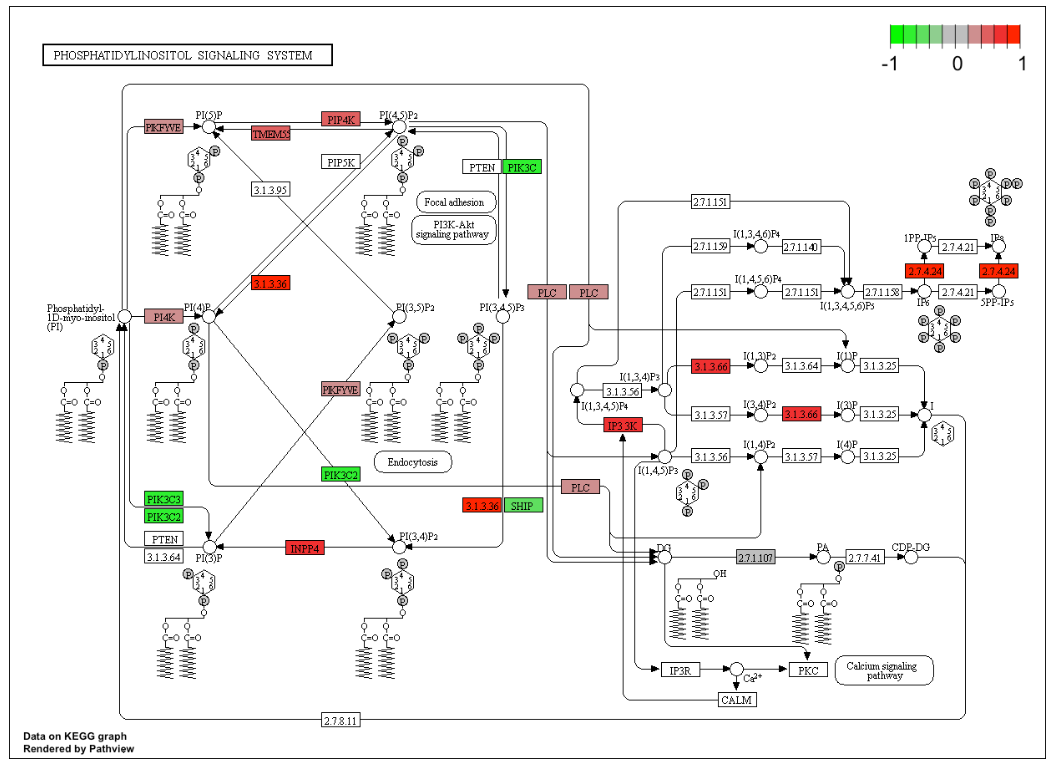
11. 通路可视化
Pathview 是一个包,它可以获取显著差异表达基因的 KEGG 标识符,还可以与 KEGG 数据库中发现的其他生物一起使用,并且可以绘制特定生物的任何 KEGG 途径。
# 加载包
biocLite("pathview") ; library(pathview)
# 可视化通路 (用 fold change)
## pathway.id : KEGG pathway identifier
pathview(gene.data = gene_matrix,
pathway.id = "04070",
species = "mouse")
欢迎Star -> 学习目录
国内链接 -> 学习目录
参考资料
Source: https://github.com/twbattaglia/RNAseq-workflow
本文由 mdnice 多平台发布
相关内容
热门资讯
埃菲尔铁塔在哪 中国仿建埃菲尔...
2019年4月26日,广西南宁市,街头惊现一座巨型山寨版埃菲尔铁塔,高约20米,白色塔身,造型逼真,...
苗族的传统节日 贵州苗族节日有...
【岜沙苗族芦笙节】岜沙,苗语叫“分送”,距从江县城7.5公里,是世界上最崇拜树木并以树为神的枪手部落...
北京的名胜古迹 北京最著名的景...
北京从元代开始,逐渐走上帝国首都的道路,先是成为大辽朝五大首都之一的南京城,随着金灭辽,金代从海陵王...
应用未安装解决办法 平板应用未...
---IT小技术,每天Get一个小技能!一、前言描述苹果IPad2居然不能安装怎么办?与此IPad不...
脚上的穴位图 脚面经络图对应的...
人体穴位作用图解大全更清晰直观的标注了各个人体穴位的作用,包括头部穴位图、胸部穴位图、背部穴位图、胳...
长白山自助游攻略 吉林长白山游...
昨天介绍了西坡的景点详细请看链接:一个人的旅行,据说能看到长白山天池全凭运气,您的运气如何?今日介绍...
猫咪吃了塑料袋怎么办 猫咪误食...
你知道吗?塑料袋放久了会长猫哦!要说猫咪对塑料袋的喜爱程度完完全全可以媲美纸箱家里只要一有塑料袋的响...
demo什么意思 demo版本...
618快到了,各位的小金库大概也在准备开闸放水了吧。没有小金库的,也该向老婆撒娇卖萌服个软了,一切只...
世界上最漂亮的人 世界上最漂亮...
此前在某网上,选出了全球265万颜值姣好的女性。从这些数量庞大的女性群体中,人们投票选出了心目中最美...
猫咪吃了塑料袋怎么办 猫咪误食...
你知道吗?塑料袋放久了会长猫哦!要说猫咪对塑料袋的喜爱程度完完全全可以媲美纸箱家里只要一有塑料袋的响...
埃菲尔铁塔在哪 中国仿建埃菲尔...
2019年4月26日,广西南宁市,街头惊现一座巨型山寨版埃菲尔铁塔,高约20米,白色塔身,造型逼真,...
苗族的传统节日 贵州苗族节日有...
【岜沙苗族芦笙节】岜沙,苗语叫“分送”,距从江县城7.5公里,是世界上最崇拜树木并以树为神的枪手部落...
北京的名胜古迹 北京最著名的景...
北京从元代开始,逐渐走上帝国首都的道路,先是成为大辽朝五大首都之一的南京城,随着金灭辽,金代从海陵王...